本章是一个完整的微生物组数据分析流程的代码实现,主要基于R语言完成。Python的部分工具也可以在特定步骤中使用(如功能预测和网络分析)。为了简化演示,我们将使用公开的16S rRNA测序数据集(如mockrobiota或QIIME2教程中的示例数据),并假设数据已经下载到本地。
数据准备与预处理
样品信息整理
# 加载必要的包 library (tidyverse)# 示例元数据文件(CSV格式) <- read_tsv ("data/MiSeq_SOP/mouse.time.design" ) # 假设包含采样时间、地点、重复等信息 head (metadata)
# A tibble: 6 × 2
group time
<chr> <chr>
1 F3D0 Early
2 F3D1 Early
3 F3D141 Late
4 F3D142 Late
5 F3D143 Late
6 F3D144 Late
原始数据导入与质控
# 安装并加载FastQC和Cutadapt # system("fastqc raw_data/*.fastq.gz -o fastqc_results/") # 运行FastQC生成质量报告 # system("cutadapt -a ADAPTER_SEQUENCE -o trimmed_data/sample.fastq raw_data/sample.fastq") # 使用Cutadapt去除接头 # R中读取FASTQ文件(可选) library (ShortRead)<- readFastq ("data/MiSeq_SOP/F3D0_S188_L001_R1_001.fastq.gz" )summary (fastq_data)
Length Class Mode
7793 ShortReadQ S4
ASV构建
序列去噪与错误校正
在开始ASV构建之前,我们首先需要设置输入文件路径并读取数据。这里我们使用DADA2包来处理双端测序数据。
# 使用DADA2进行ASV构建 library (dada2)# 设置路径 <- "data/MiSeq_SOP" <- sort (list.files (path, pattern = "_R1_001.fastq.gz" ))<- sort (list.files (path, pattern = "_R2_001.fastq.gz" ))<- sapply (strsplit (fnFs, "_" ), ` [ ` , 1 )
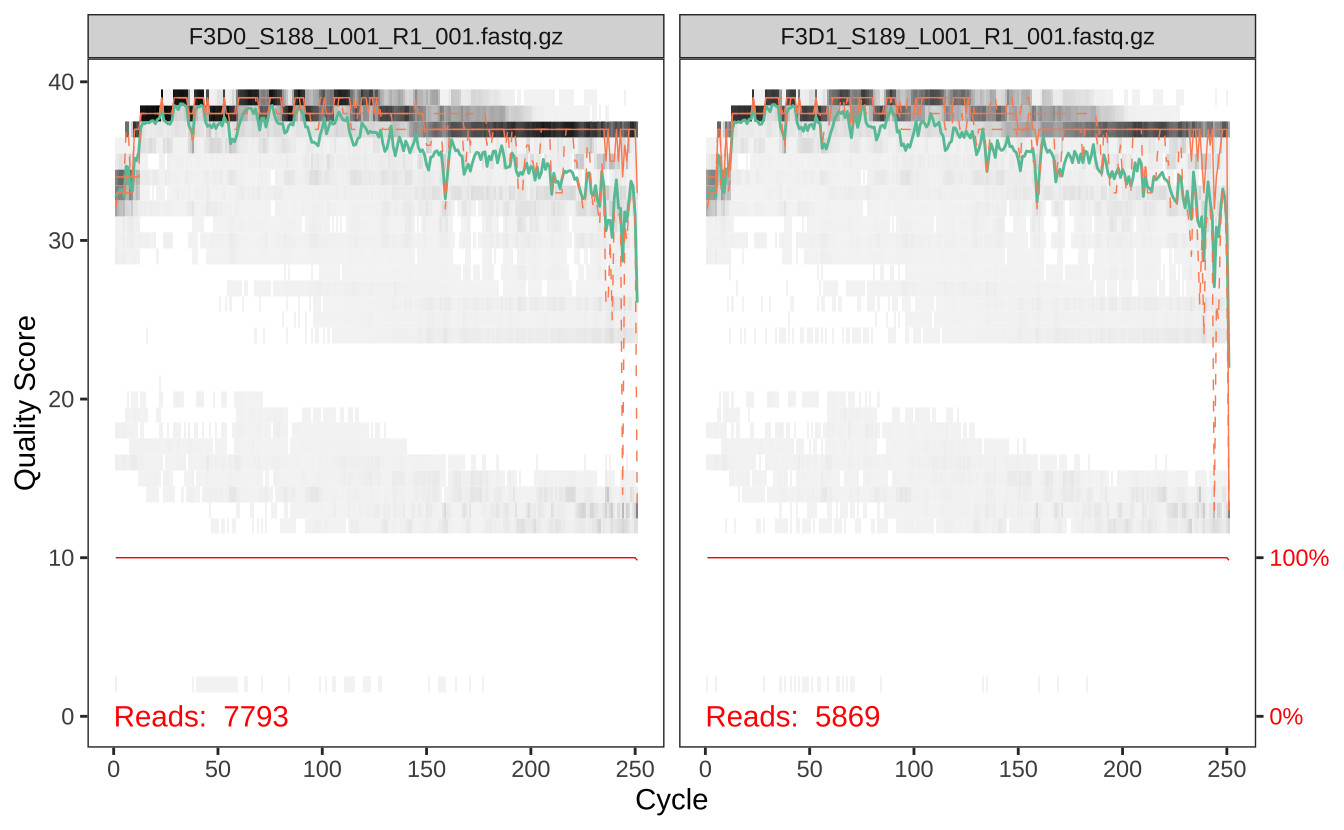
接下来我们需要评估测序数据的质量。通过查看正向读段的质量分布图,我们可以确定合适的质量过滤参数。
# 查看正向读段的质量分布 plotQualityProfile (file.path (path, fnFs[1 : 2 ])) # 可视化前两个样本的正向读段质量分布
从质量分布图中,我们可以看到碱基质量随着测序位置的变化趋势。通常在读段末端质量会下降,这有助于我们确定截断位置。
同样,我们也需要查看反向读段的质量分布。
# 查看反向读段的质量分布 plotQualityProfile (file.path (path, fnRs[1 : 2 ])) # 可视化前两个样本的反向读段质量分布
根据质量分布图,我们可以看到反向读段的质量通常低于正向读段,这是正常现象。这些信息将帮助我们在下一步设置适当的质量过滤参数。
# 质量过滤 <- file.path (path, "filtered" )if (! file.exists (filt_path)) dir.create (filt_path)<- file.path (filt_path, paste0 (sample.names, "_F_filt.fastq.gz" ))<- file.path (filt_path, paste0 (sample.names, "_R_filt.fastq.gz" ))# 设置文件名 names (fnFs.filt) <- sample.namesnames (fnRs.filt) <- sample.names
# 过滤和修剪 <- filterAndTrim (file.path (path, fnFs), fnFs.filt,file.path (path, fnRs), fnRs.filt,truncLen = c (240 , 160 ), maxN = 0 , maxEE = c (2 , 2 ), truncQ = 2 , rm.phix = TRUE )head (out)
reads.in reads.out
F3D0_S188_L001_R1_001.fastq.gz 7793 7113
F3D1_S189_L001_R1_001.fastq.gz 5869 5299
F3D141_S207_L001_R1_001.fastq.gz 5958 5463
F3D142_S208_L001_R1_001.fastq.gz 3183 2914
F3D143_S209_L001_R1_001.fastq.gz 3178 2941
F3D144_S210_L001_R1_001.fastq.gz 4827 4312
# 学习错误模型 <- learnErrors (fnFs.filt, multithread = TRUE )
33514080 total bases in 139642 reads from 20 samples will be used for learning the error rates.
<- learnErrors (fnRs.filt, multithread = TRUE )
22342720 total bases in 139642 reads from 20 samples will be used for learning the error rates.
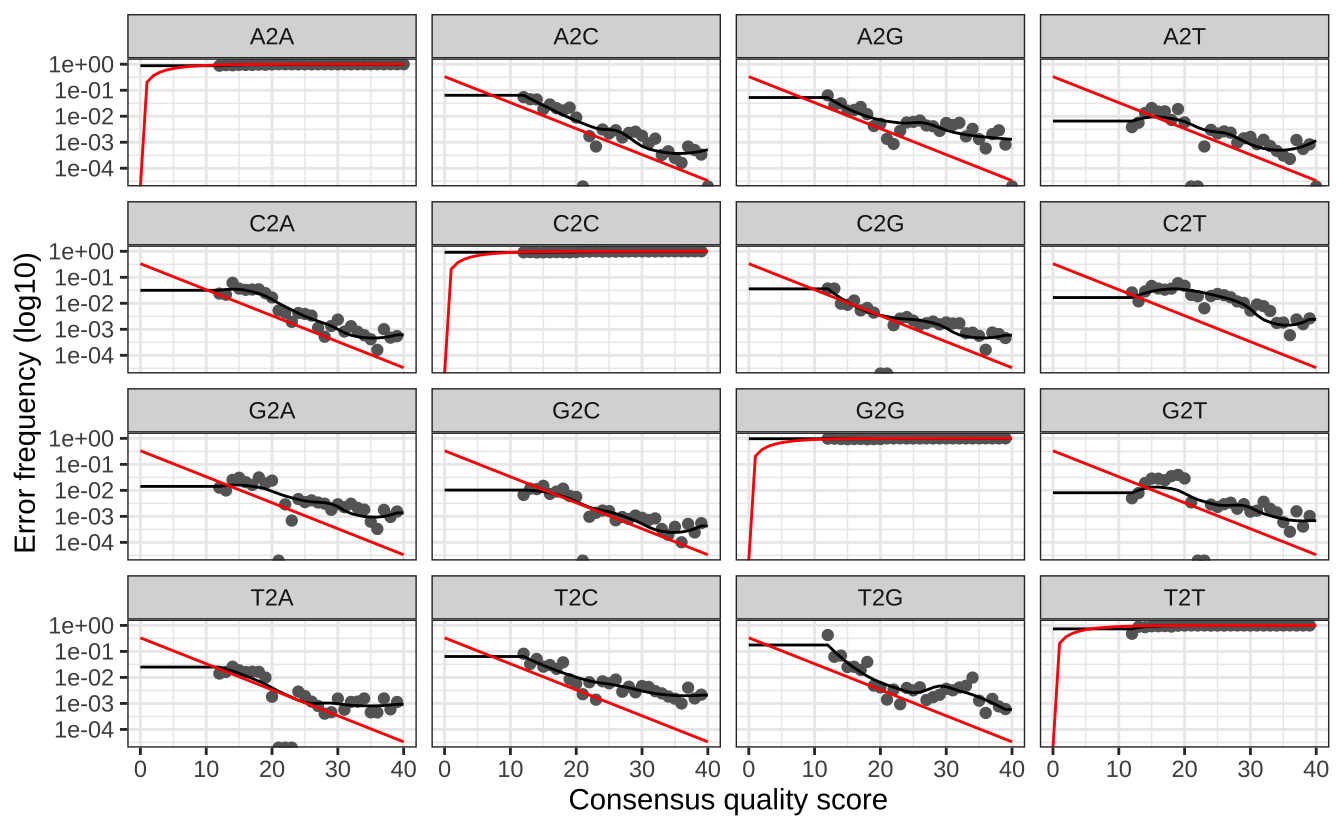
# 可视化错误模型 plotErrors (errF, nominalQ = TRUE )plotErrors (errR, nominalQ = TRUE )
# 使用DADA2进行序列去噪 <- dada (fnFs.filt, err = errF, multithread = TRUE )
Sample 1 - 7113 reads in 1979 unique sequences.
Sample 2 - 5299 reads in 1639 unique sequences.
Sample 3 - 5463 reads in 1477 unique sequences.
Sample 4 - 2914 reads in 904 unique sequences.
Sample 5 - 2941 reads in 939 unique sequences.
Sample 6 - 4312 reads in 1267 unique sequences.
Sample 7 - 6741 reads in 1756 unique sequences.
Sample 8 - 4560 reads in 1438 unique sequences.
Sample 9 - 15637 reads in 3590 unique sequences.
Sample 10 - 11413 reads in 2762 unique sequences.
Sample 11 - 12017 reads in 3021 unique sequences.
Sample 12 - 5032 reads in 1566 unique sequences.
Sample 13 - 18075 reads in 3707 unique sequences.
Sample 14 - 6250 reads in 1479 unique sequences.
Sample 15 - 4052 reads in 1195 unique sequences.
Sample 16 - 7369 reads in 1832 unique sequences.
Sample 17 - 4765 reads in 1183 unique sequences.
Sample 18 - 4871 reads in 1382 unique sequences.
Sample 19 - 6504 reads in 1709 unique sequences.
Sample 20 - 4314 reads in 897 unique sequences.
<- dada (fnRs.filt, err = errR, multithread = TRUE )
Sample 1 - 7113 reads in 1660 unique sequences.
Sample 2 - 5299 reads in 1349 unique sequences.
Sample 3 - 5463 reads in 1335 unique sequences.
Sample 4 - 2914 reads in 853 unique sequences.
Sample 5 - 2941 reads in 880 unique sequences.
Sample 6 - 4312 reads in 1286 unique sequences.
Sample 7 - 6741 reads in 1803 unique sequences.
Sample 8 - 4560 reads in 1265 unique sequences.
Sample 9 - 15637 reads in 3414 unique sequences.
Sample 10 - 11413 reads in 2522 unique sequences.
Sample 11 - 12017 reads in 2771 unique sequences.
Sample 12 - 5032 reads in 1415 unique sequences.
Sample 13 - 18075 reads in 3290 unique sequences.
Sample 14 - 6250 reads in 1390 unique sequences.
Sample 15 - 4052 reads in 1134 unique sequences.
Sample 16 - 7369 reads in 1635 unique sequences.
Sample 17 - 4765 reads in 1084 unique sequences.
Sample 18 - 4871 reads in 1161 unique sequences.
Sample 19 - 6504 reads in 1502 unique sequences.
Sample 20 - 4314 reads in 732 unique sequences.
# 查看去噪结果 #head(dadaFs[[1]]) #head(dadaRs[[1]])
# 合并配对末端 <- mergePairs (dadaFs, fnFs.filt, dadaRs, fnRs.filt)# 查看合并结果 head (mergers[[1 ]])
sequence
1 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGCAGGCGGAAGATCAAGTCAGCGGTAAAATTGAGAGGCTCAACCTCTTCGAGCCGTTGAAACTGGTTTTCTTGAGTGAGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCTCAACTGACGCTCATGCACGAAAGTGTGGGTATCGAACAGG
2 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGCCTGCCAAGTCAGCGGTAAAATTGCGGGGCTCAACCCCGTACAGCCGTTGAAACTGCCGGGCTCGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACCCCGATTGCGAAGGCAGCATACCGGCGCCCTACTGACGCTGAGGCACGAAAGTGCGGGGATCAAACAGG
3 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGCTGTTAAGTCAGCGGTCAAATGTCGGGGCTCAACCCCGGCCTGCCGTTGAAACTGGCGGCCTCGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCCCGACTGACGCTGAGGCACGAAAGCGTGGGTATCGAACAGG
4 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGCTTTTAAGTCAGCGGTAAAAATTCGGGGCTCAACCCCGTCCGGCCGTTGAAACTGGGGGCCTTGAGTGGGCGAGAAGAAGGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACCCCGATTGCGAAGGCAGCCTTCCGGCGCCCTACTGACGCTGAGGCACGAAAGTGCGGGGATCGAACAGG
5 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGCAGGCGGACTCTCAAGTCAGCGGTCAAATCGCGGGGCTCAACCCCGTTCCGCCGTTGAAACTGGGAGCCTTGAGTGCGCGAGAAGTAGGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCCTACCGGCGCGCAACTGACGCTCATGCACGAAAGCGTGGGTATCGAACAGG
6 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGATGCCAAGTCAGCGGTAAAAAAGCGGTGCTCAACGCCGTCGAGCCGTTGAAACTGGCGTTCTTGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCCCTACTGACGCTGAGGCACGAAAGCGTGGGTATCGAACAGG
abundance forward reverse nmatch nmismatch nindel prefer accept
1 579 1 1 148 0 0 1 TRUE
2 470 2 2 148 0 0 2 TRUE
3 449 3 4 148 0 0 1 TRUE
4 430 4 3 148 0 0 2 TRUE
5 345 5 6 148 0 0 1 TRUE
6 282 6 5 148 0 0 2 TRUE
# 构建ASV表 <- makeSequenceTable (mergers)dim (seqtab)
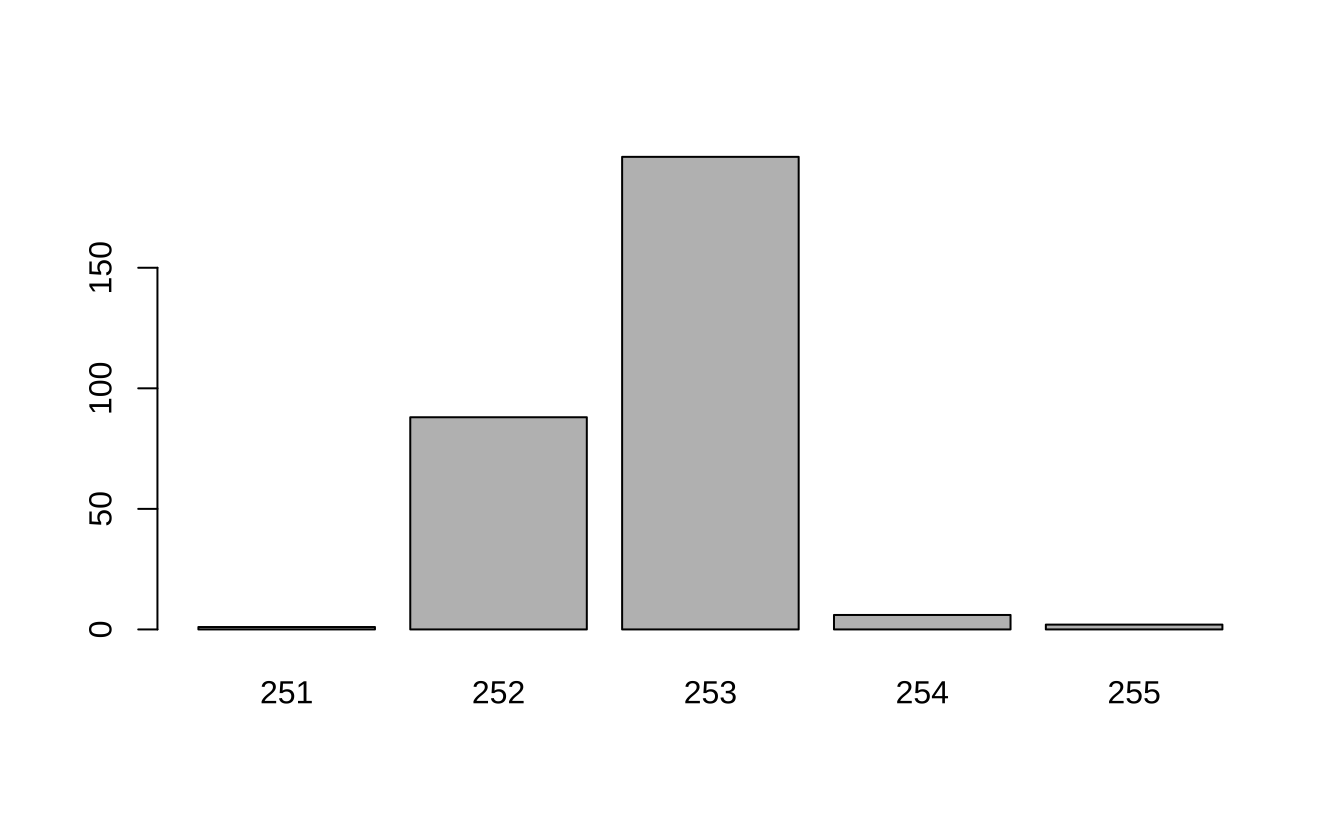
# 查看序列长度分布 table (nchar (getSequences (seqtab))) |> barplot ()
# 去除嵌合体 <- removeBimeraDenovo (seqtab, method = "consensus" , multithread = TRUE )# 计算嵌合体去除效率 sum (seqtab.nochim)/ sum (seqtab)
序列数量统计
# 计算每个步骤的序列数量 <- function (x) sum (getUniques (x))<- cbind (out, sapply (dadaFs, getN), sapply (dadaRs, getN), sapply (mergers, getN), rowSums (seqtab.nochim))# If processing a single sample, remove the sapply calls: e.g. replace sapply(dadaFs, getN) with getN(dadaFs) colnames (track) <- c ("input" , "filtered" , "denoisedF" , "denoisedR" , "merged" , "nonchim" )rownames (track) <- sample.nameshead (track)
input filtered denoisedF denoisedR merged nonchim
F3D0 7793 7113 6976 6979 6540 6528
F3D1 5869 5299 5227 5239 5028 5017
F3D141 5958 5463 5331 5357 4986 4863
F3D142 3183 2914 2799 2830 2595 2521
F3D143 3178 2941 2822 2868 2553 2519
F3D144 4827 4312 4151 4228 3646 3507
分类学注释
使用SILVA数据库进行分类学注释
在进行物种分类注释之前,我们需要准备SILVA参考数据库。这个数据库包含了已知细菌和古菌的16S rRNA基因序列及其分类信息。
# 下载SILVA数据库(如果尚未下载) # system("wget https://zenodo.org/records/14169026/files/silva_nr99_v138.2_toSpecies_trainset.fa.gz") = "./data/silva_v138.2/silva_nr99_v138.2_wSpecies_train_set.fa.gz" # 分类学注释 <- assignTaxonomy (seqtab.nochim, silva_db, multithread = TRUE )
注释完成后,我们可以查看注释结果。结果中包含了每个ASV的完整分类学信息,从门到种水平。
# 查看分类学注释结果 <- taxa # Removing sequence rownames for display only rownames (taxa.print) <- NULL head (taxa.print)
Kingdom Phylum Class Order Family
[1,] "Bacteria" "Bacteroidota" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
[2,] "Bacteria" "Bacteroidota" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
[3,] "Bacteria" "Bacteroidota" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
[4,] "Bacteria" "Bacteroidota" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
[5,] "Bacteria" "Bacteroidota" "Bacteroidia" "Bacteroidales" "Bacteroidaceae"
[6,] "Bacteria" "Bacteroidota" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
Genus Species
[1,] NA NA
[2,] NA NA
[3,] NA NA
[4,] NA NA
[5,] "Bacteroides" "caecimuris"
[6,] NA NA
从输出结果可以看到,每个ASV都被赋予了一个完整的分类系统,包括门(Phylum)、纲(Class)、目(Order)、科(Family)、属(Genus)和种(Species)水平的分类信息。如果某一分类级别无法确定,则会显示为NA。
创建phyloseq对象
library (phyloseq)<- rownames (seqtab.nochim)<- sapply (strsplit (samples.out, "D" ), ` [ ` , 1 )<- substr (subject,1 ,1 )<- substr (subject,2 ,999 )<- as.integer (sapply (strsplit (samples.out, "D" ), ` [ ` , 2 ))<- data.frame (Subject= subject, Gender= gender, Day= day)$ When <- "Early" $ When[samdf$ Day> 100 ] <- "Late" rownames (samdf) <- samples.out
<- phyloseq (otu_table (seqtab.nochim, taxa_are_rows= FALSE ), sample_data (samdf), tax_table (taxa))<- prune_samples (sample_names (ps) != "Mock" , ps) # Remove mock sample
# 添加序列名称 <- Biostrings:: DNAStringSet (taxa_names (ps))names (dna) <- taxa_names (ps)<- merge_phyloseq (ps, dna)taxa_names (ps) <- paste0 ("ASV" , seq (ntaxa (ps)))
phyloseq-class experiment-level object
otu_table() OTU Table: [ 232 taxa and 19 samples ]
sample_data() Sample Data: [ 19 samples by 4 sample variables ]
tax_table() Taxonomy Table: [ 232 taxa by 7 taxonomic ranks ]
refseq() DNAStringSet: [ 232 reference sequences ]
多样性分析
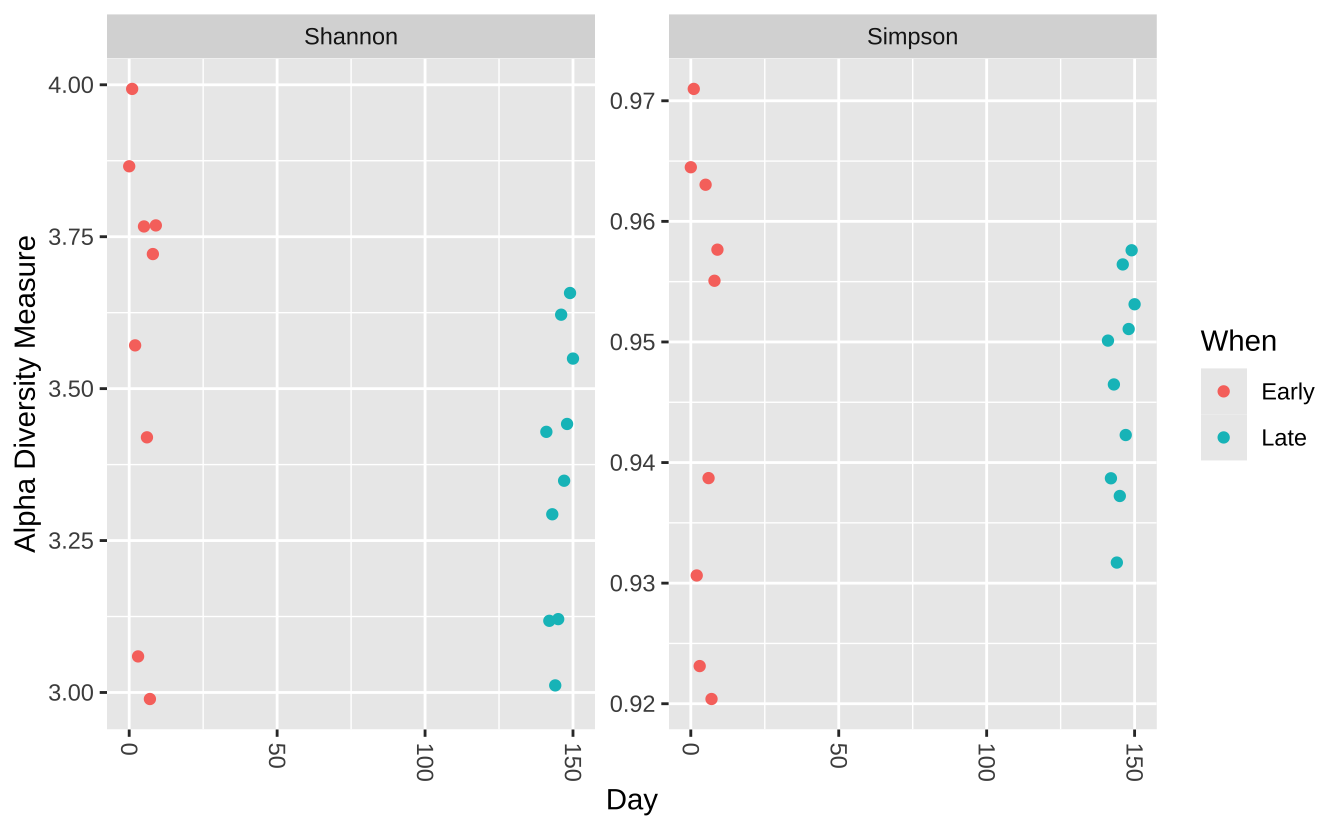
Alpha多样性分析
plot_richness (ps, x= "Day" , measures= c ("Shannon" , "Simpson" ), color= "When" )
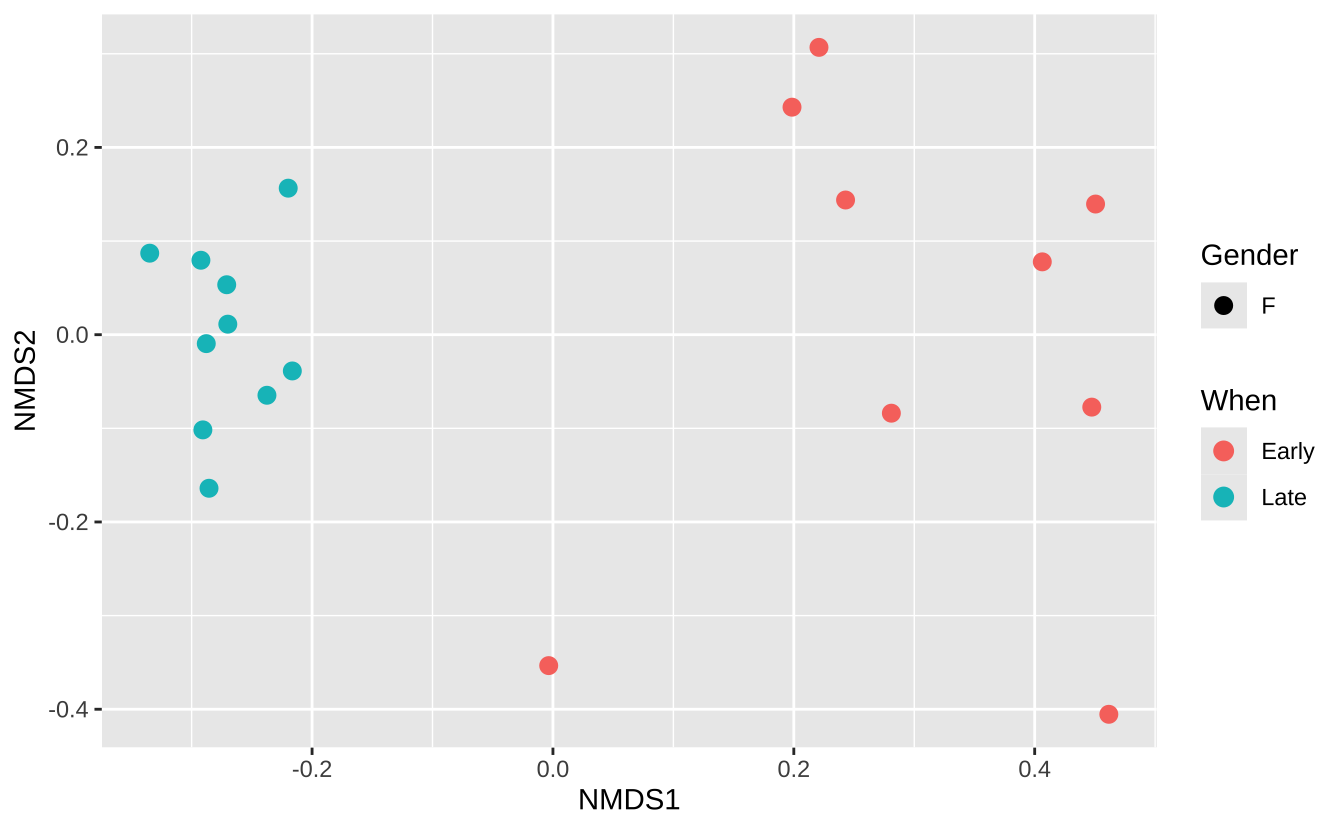
Beta多样性分析
# 计算Bray-Curtis距离 <- transform_sample_counts (ps, function (otu) otu/ sum (otu))<- ordinate (ps.prop, method= "NMDS" , distance= "bray" )
Run 0 stress 0.08043117
Run 1 stress 0.08076338
... Procrustes: rmse 0.01052743 max resid 0.03240295
Run 2 stress 0.08076337
... Procrustes: rmse 0.01048205 max resid 0.03225559
Run 3 stress 0.08076338
... Procrustes: rmse 0.01053409 max resid 0.03242571
Run 4 stress 0.1326152
Run 5 stress 0.08076338
... Procrustes: rmse 0.01054091 max resid 0.03244821
Run 6 stress 0.08076337
... Procrustes: rmse 0.01049199 max resid 0.03228863
Run 7 stress 0.08616061
Run 8 stress 0.08043116
... New best solution
... Procrustes: rmse 1.226092e-06 max resid 2.909654e-06
... Similar to previous best
Run 9 stress 0.08616061
Run 10 stress 0.08076336
... Procrustes: rmse 0.01047795 max resid 0.03224207
Run 11 stress 0.08076337
... Procrustes: rmse 0.01052241 max resid 0.03238733
Run 12 stress 0.09477093
Run 13 stress 0.101063
Run 14 stress 0.08616061
Run 15 stress 0.08043116
... Procrustes: rmse 4.902388e-07 max resid 9.732552e-07
... Similar to previous best
Run 16 stress 0.08616061
Run 17 stress 0.08076341
... Procrustes: rmse 0.01057329 max resid 0.03255292
Run 18 stress 0.08076343
... Procrustes: rmse 0.01059541 max resid 0.03262505
Run 19 stress 0.1358145
Run 20 stress 0.1274324
*** Best solution repeated 2 times
# 绘制NMDS图 plot_ordination (ps.prop, ord.nmds.bray, color = "When" , shape = "Gender" ) + geom_point (size = 3 )
差异分析
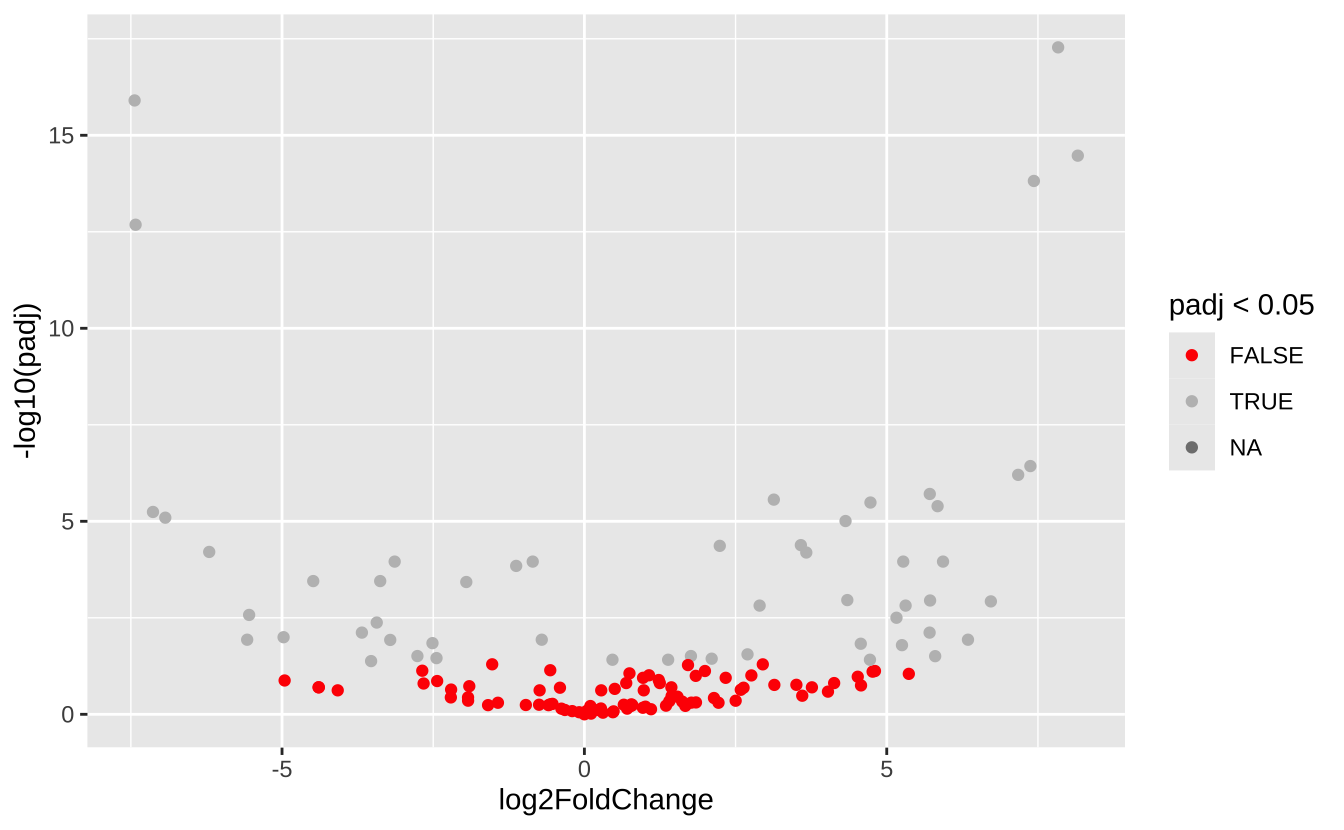
差异丰度分析
library (DESeq2)# 准备DESeq2输入数据 <- phyloseq_to_deseq2 (ps, ~ When)<- DESeq (dds)# 获取差异显著的ASV <- results (dds, contrast = c ("When" , "Early" , "Late" ))<- res[which (res$ padj < 0.05 ), ]head (sig_asvs)
log2 fold change (MLE): When Early vs Late
Wald test p-value: When Early vs Late
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue padj
<numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
ASV4 368.082 -1.127928 0.266729 -4.22875 2.34994e-05 0.000143040
ASV6 240.045 -3.137408 0.730308 -4.29601 1.73902e-05 0.000110665
ASV7 250.823 -0.854399 0.198571 -4.30274 1.68698e-05 0.000110665
ASV10 181.772 0.464222 0.190101 2.44198 1.46071e-02 0.038584923
ASV11 136.812 -1.952974 0.490931 -3.97811 6.94666e-05 0.000374051
ASV12 115.139 -3.210652 1.098399 -2.92303 3.46643e-03 0.011836602
# 绘制火山图 ggplot (res, aes (x = log2FoldChange, y = - log10 (padj))) + geom_point (aes (color = padj < 0.05 )) + scale_color_manual (values = c ("red" , "grey" ))
代谢功能预测
使用PICRUSt2进行功能预测
# 在命令行运行PICRUSt2 picrust2_pipeline.py -s seqtab_nochim.fasta -i seqtab_nochim.biom -o picrust2_output --threads 4
功能差异分析
# 加载KEGG通路预测结果 <- read.table ("picrust2_output/path_abun.tsv" , header = TRUE , row.names = 1 )<- DESeq2:: DESeqDataSetFromMatrix (countData = kegg_pathways, colData = metadata, design = ~ TimePoint)
共现网络分析
在R中分析微生物数据时,构建共现网络(Co-occurrence Network)是一种常见的方法,用于探索不同微生物群落之间的相互关系。以下是详细的步骤和代码示例,帮助您从头到尾完成这一任务。
数据准备
# 去除低丰度OTU(例如总丰度小于10的OTU) <- prune_taxa (taxa_sums (ps) > 30 , ps)<- otu_table (ps) |> as.data.frame ()# 检查是否有缺失值 if (any (is.na (otu_table))) {stop ("OTU表中存在缺失值,请先处理缺失值!" )
计算相关性矩阵
共现网络通常基于微生物之间的相关性(如Spearman、Pearson或Kendall相关系数)。您可以使用 cor() 函数计算相关性矩阵。
library (Hmisc)# 计算Spearman相关性矩阵 <- rcorr (as.matrix (otu_table), type = "spearman" )str (cor_matrix)
List of 4
$ r : num [1:153, 1:153] 1 0.909 0.881 0.575 0.489 ...
..- attr(*, "dimnames")=List of 2
.. ..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
.. ..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
$ n : int [1:153, 1:153] 19 19 19 19 19 19 19 19 19 19 ...
..- attr(*, "dimnames")=List of 2
.. ..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
.. ..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
$ P : num [1:153, 1:153] NA 7.32e-08 6.49e-07 9.94e-03 3.34e-02 ...
..- attr(*, "dimnames")=List of 2
.. ..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
.. ..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
$ type: chr "spearman"
- attr(*, "class")= chr "rcorr"
筛选显著的相关性
为了减少噪声,通常只保留显著的相关性(例如p值小于某个阈值)。可以使用 Hmisc 包中的 rcorr() 函数来计算相关性和p值。
# 计算相关性和p值 <- rcorr (as.matrix (otu_table), type = "spearman" )<- cor_test$ r # 相关性矩阵 <- cor_test$ P # p值矩阵 # 设置显著性阈值 <- 0.05 <- ifelse (p_values < p_threshold, cor_values, 0 )str (significant_cor)
num [1:153, 1:153] NA 0.909 0.881 0.575 0.489 ...
- attr(*, "dimnames")=List of 2
..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
构建邻接矩阵
将显著的相关性转换为邻接矩阵(Adjacency Matrix),以便后续用于网络分析。可以设置一个相关性阈值(例如绝对值大于0.6)来进一步筛选强相关性。
# 设置相关性阈值 <- 0.8 <- ifelse (abs (significant_cor) > cor_threshold, significant_cor, 0 )diag (adj_matrix) <- 0 # 对角线设为0,避免自相关 str (adj_matrix)
num [1:153, 1:153] 0 0.909 0.881 0 0 ...
- attr(*, "dimnames")=List of 2
..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
..$ : chr [1:153] "ASV1" "ASV2" "ASV3" "ASV4" ...
可视化共现网络
使用 igraph 或 networkD3 等包可以可视化共现网络。
使用 igraph 包
# 加载igraph包 library (igraph)# 将邻接矩阵转换为图对象 <- graph_from_adjacency_matrix (adj_matrix, mode = "undirected" , weighted = TRUE )# 绘制网络图 plot (graph,vertex.size = 10 ,vertex.color = "lightblue" ,vertex.label.color = "black" ,edge.width = E (graph)$ weight * 5 , # 边权重影响宽度 main = "Microbiome Co-occurrence Network" )
进一步分析
模块检测 :可以使用 igraph 中的社区检测算法(如Louvain方法)来识别网络中的模块。网络属性 :计算网络的拓扑属性(如节点度、聚类系数等)以深入了解微生物群落的结构。
# 检测社区 <- cluster_louvain (graph)<- membership (communities)print (membership)# 计算节点度 <- degree (graph)print (degrees)以上代码提供了一个完整的微生物组数据分析流程框架,具体细节可以根据实际数据和研究需求调整。